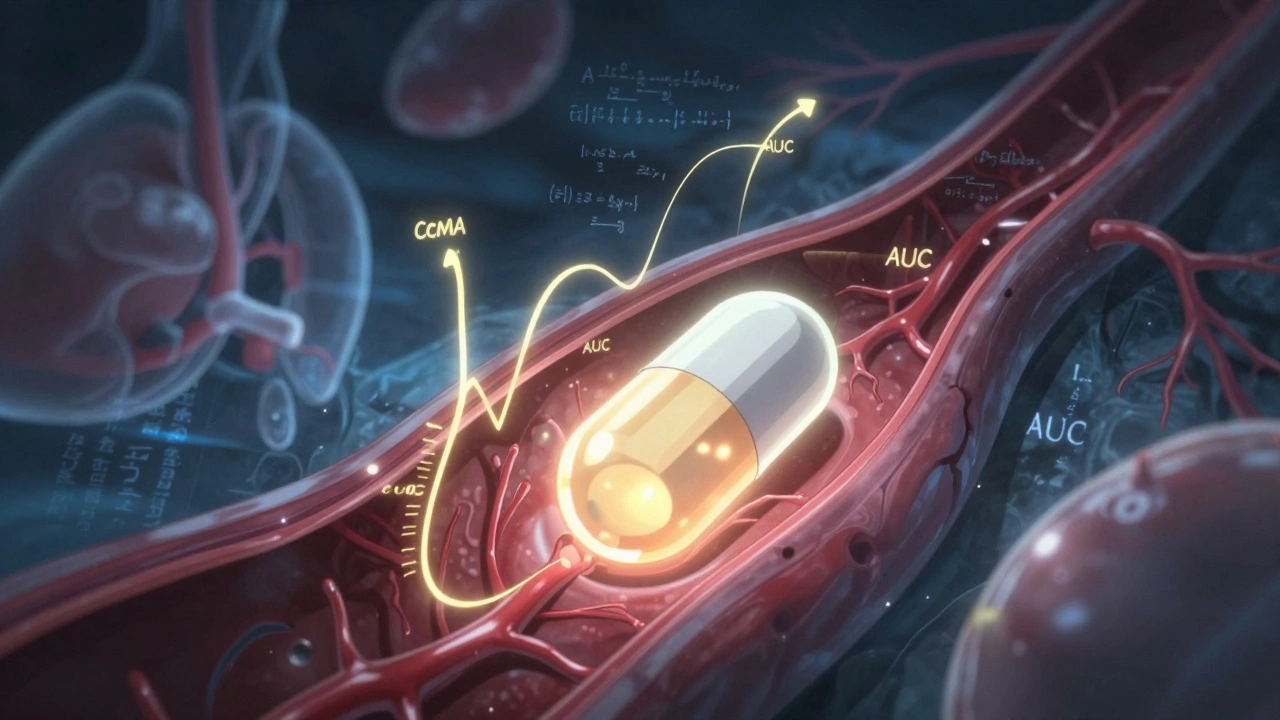
Quand un médicament générique arrive sur le marché, comment sait-on qu’il fonctionne exactement comme le médicament d’origine ? La réponse se trouve dans les études pharmacocinétiques. Ce ne sont pas des tests de laboratoire simples, ni des comparaisons de composition. Ce sont des études menées sur des êtres humains, avec des prises de sang, des courbes de concentration dans le sang, et des calculs statistiques stricts. Et pourtant, malgré leur rôle central, ces études ne sont pas un « or » pur - elles sont un outil, puissant mais limité.
Comment ça marche, une étude de bioéquivalence ?
Imaginez que vous prenez un comprimé. Ce comprimé doit se dissoudre dans votre estomac, traverser votre paroi intestinale, entrer dans votre sang, puis atteindre sa cible - un organe, une cellule, un récepteur. La vitesse et la quantité de médicament qui arrivent dans votre sang sont ce qu’on appelle la pharmacocinétique. Deux médicaments sont dits bioéquivalents si, après administration dans la même dose, ils produisent des concentrations similaires dans le sang, à un très faible écart près. Les deux paramètres clés mesurés sont : Cmax (la concentration maximale atteinte dans le sang) et AUC (l’aire sous la courbe, qui représente la quantité totale de médicament absorbée dans le temps). Pour qu’un générique soit approuvé, la moyenne géométrique des rapports entre le générique et le médicament d’origine doit se situer entre 80 % et 125 % pour ces deux paramètres. C’est la règle imposée par la FDA aux États-Unis, l’EMA en Europe, et adoptée par plus de 50 agences nationales à travers le monde. Ces études se font sur 24 à 36 volontaires en bonne santé, dans un design en croisement : chaque participant prend d’abord le générique, puis après une période de lavage, le médicament d’origine. Les prises de sang sont faites toutes les 15 à 30 minutes pendant 24 à 72 heures. Les analyses statistiques utilisent l’ANOVA pour s’assurer que les différences observées ne sont pas dues au hasard.Pourquoi cette plage de 80-125 % ?
Ce n’est pas un chiffre tiré au hasard. C’est le résultat de décennies d’analyse clinique. Des études ont montré que si la concentration dans le sang varie de moins de 20 % entre deux formulations, les effets thérapeutiques sont pratiquement identiques. Pour la plupart des médicaments, c’est suffisant. Mais pas pour tous. Pour les médicaments à indice thérapeutique étroit - comme la warfarine, la phénytoïne ou la digoxine - la marge est resserrée. Là, les autorités exigent une plage de 90 % à 111 %. Pourquoi ? Parce que même une petite variation peut entraîner un caillot sanguin ou une hémorragie. Un générique qui passe la norme classique pourrait échouer ici. Et c’est pourquoi la FDA a publié des guides spécifiques pour 28 de ces médicaments en 2023.Les limites invisibles de la pharmacocinétique
Ce n’est pas parce qu’un générique a la même concentration dans le sang qu’il agit exactement comme le médicament d’origine. La pharmacocinétique mesure ce qui passe dans le sang. Elle ne mesure pas ce qui se passe dans la peau, les poumons, le cerveau, ou les articulations. Prenons les crèmes ou les pommades. Pour les médicaments topiques, mesurer la concentration dans le sang est inutile - le médicament ne doit pas entrer dans la circulation générale. Il doit agir localement. Dans ces cas, les études pharmacocinétiques classiques échouent. Des méthodes alternatives comme les tests de perméation in vitro (IVPT) avec peau humaine congelée, ou la dermatopharmacocinétique (DMD), sont devenues plus fiables. Une étude de 2014 a montré que ces méthodes étaient plus précises et moins variables que des essais cliniques avec plus de 500 patients. Même pour les comprimés, des cas étranges ont été observés. En 2010, une étude publiée dans PLOS ONE a révélé que deux génériques de gentamicine - avec la même formule, la même pureté, et des tests in vitro identiques - avaient des profils thérapeutiques très différents chez les patients. Le médicament était « pharmaceutiquement équivalent » mais pas « thérapeutiquement équivalent ». La pharmacocinétique n’a pas pu le prédire.
Coûts, délais, et défis pour les fabricants
Faire une étude de bioéquivalence, ce n’est pas bon marché. Entre 300 000 et 1 million de dollars. Et ça prend entre 12 et 18 mois. Pour une petite entreprise, c’est une barrière énorme. Les formulations modifiées - à libération prolongée, à libération ciblée, ou avec des excipients complexes - sont particulièrement difficiles. Un changement minime dans un additif peut altérer la vitesse de dissolution, même si la molécule active est identique. La FDA a répondu en publiant 1 857 guides spécifiques à chaque produit en 2023. Chaque médicament a ses propres règles. Ce n’est plus une approche « une taille unique ». C’est une carte routière complexe, avec des exigences différentes selon le type de comprimé, la voie d’administration, ou la maladie traitée.La révolution des modèles informatiques
Une nouvelle génération d’outils émerge : les modèles pharmacocinétiques basés sur la physiologie (PBPK). Ces logiciels simulent comment un médicament va se comporter dans le corps humain - en tenant compte de l’âge, du poids, du pH gastrique, de la perméabilité intestinale, et même du microbiote. Depuis 2020, la FDA accepte ces modèles pour accorder des dérogations aux études in vivo pour certains médicaments de la classe BCS I (très solubles et très perméables). C’est une révolution. Pour 15 % des médicaments, les fabricants n’ont plus besoin de recruter des volontaires. Ils peuvent prouver l’équivalence avec un ordinateur. Cela réduit les coûts, accélère les délais, et évite les risques liés aux essais humains.
Le vrai critère : la sécurité et l’efficacité du patient
L’Organisation mondiale de la santé (OMS) définit l’équivalence thérapeutique comme la capacité de deux médicaments à produire « des effets similaires en termes d’efficacité et de sécurité ». La pharmacocinétique est un moyen, pas la fin. Elle est fiable pour 95 % des génériques approuvés par la FDA en 2022. Mais elle ne peut pas tout prédire. Les agences réglementaires savent cela. C’est pourquoi elles utilisent un mélange de méthodes : dissolution in vitro, études pharmacocinétiques, études pharmacodynamiques, et même des essais cliniques pour les produits complexes. Le vrai « or » n’est pas la méthode, c’est le résultat : un patient qui reçoit un générique et qui n’a pas de réaction inattendue, pas de perte d’efficacité, pas d’effet secondaire nouveau. Les études pharmacocinétiques sont le pilier du système. Mais elles ne sont pas le seul pilier. Et ce n’est pas parce qu’une méthode est largement utilisée qu’elle est parfaite. La science progresse quand on accepte ses limites.Les alternatives qui montent
Pour certains médicaments, les tests in vitro sont devenus plus fiables que les tests sur les humains. Une étude de 2009 publiée sur PMC a montré que, pour certains comprimés à libération immédiate, des tests de dissolution bien conçus pouvaient prédire l’équivalence avec moins de variabilité que les études pharmacocinétiques. Pour les inhalateurs, les études de dépôt pulmonaire avec des modèles de simulation sont en cours de validation. Pour les médicaments injectables, les comparaisons de profils de libération sont de plus en plus utilisées. La tendance est claire : on passe d’une approche universelle à une approche personnalisée, adaptée à chaque médicament.Que faire si vous prenez un générique ?
Si vous prenez un médicament courant - un antibiotique, un antihypertenseur, un antidouleur - vous n’avez rien à craindre. Les génériques sont sûrs, efficaces, et testés rigoureusement. Mais si vous prenez un médicament à indice thérapeutique étroit - comme un anticoagulant, un anticonvulsivant, ou un traitement pour l’hypothyroïdie - parlez à votre médecin. Certains génériques peuvent nécessiter une surveillance plus fine. Votre médecin peut vérifier la concentration dans le sang ou choisir de rester sur le même générique si vous avez déjà bien répondu à celui-là. Le système est conçu pour protéger. Il n’est pas parfait. Mais il est le meilleur que nous ayons - et il continue d’évoluer.Pourquoi les études pharmacocinétiques sont-elles obligatoires pour les médicaments génériques ?
Elles sont obligatoires parce qu’elles permettent de prouver que le générique libère la même quantité de médicament dans le sang que le médicament d’origine, dans un délai similaire. Sans cette preuve, on ne peut pas garantir que le générique aura le même effet thérapeutique. C’est la seule méthode reconnue pour la majorité des comprimés et capsules orales.
Tous les génériques passent-ils ces études ?
Oui, tous les génériques destinés à être absorbés dans la circulation sanguine doivent passer des études de bioéquivalence. Les exceptions sont rares : certains produits topiques, inhalés, ou injectables peuvent utiliser d’autres méthodes validées. Mais pour les comprimés, gélules, et sirops oraux, c’est obligatoire.
Pourquoi certains génériques ne fonctionnent-ils pas aussi bien chez certains patients ?
Parce que la bioéquivalence ne garantit pas une identité parfaite. Des différences dans les excipients, la forme du comprimé, ou la vitesse de dissolution peuvent affecter l’absorption chez certaines personnes - surtout si elles ont un estomac sensible, un problème de motilité intestinale, ou prennent d’autres médicaments. Pour les médicaments à indice thérapeutique étroit, ces petites différences peuvent avoir un impact clinique.
Les modèles informatiques vont-ils remplacer les études sur les humains ?
Pour certains médicaments, oui. Les modèles PBPK sont déjà acceptés pour les médicaments très solubles et très perméables (BCS Classe I). Mais pour les formulations complexes - comme les comprimés à libération prolongée ou les produits topiques - les études humaines restent nécessaires. Les modèles sont un outil d’aide, pas une solution universelle.
Comment savoir si un générique est de bonne qualité ?
En France et en Europe, tous les génériques doivent être approuvés par l’ANSM ou l’EMA. Vous pouvez vérifier sur le site de l’ANSM si un générique est autorisé. Le prix n’est pas un indicateur de qualité. Un générique bon marché peut être aussi efficace qu’un cher - tant qu’il a passé les tests de bioéquivalence. Si vous avez un doute, parlez à votre pharmacien ou à votre médecin.


9 Commentaires
Je trouve fascinant comment on peut réduire la complexité du corps humain à deux chiffres : Cmax et AUC. On parle de vie et de mort, et on se contente d’une plage de 80 à 125 %. C’est à la fois ingénieux et un peu effrayant.
La science est belle quand elle simplifie, mais elle devient dangereuse quand elle oublie que chaque corps est un univers unique.
Ok mais sérieux?? Les génériques?? Moi j’ai eu une crise d’épilepsie après un changement de générique… et on m’a dit que c’était « dans les normes »… C’est pas normal ça!!
Je suis infirmière et je vois tous les jours des patients qui paniquent en changeant de générique… même si c’est « théoriquement » équivalent. On a besoin de plus de transparence sur les excipients, pas juste des courbes de sang.
Et puis, pourquoi on ne publie pas les données brutes ? Tous les labos les ont… mais personne ne les partage. C’est un peu comme un secret d’État.
Les modèles PBPK, c’est la révolution. J’ai travaillé sur un projet avec un labo qui a remplacé 3 études humaines par un modèle. Coût : 50k au lieu de 800k. Temps : 3 mois au lieu de 18.
La pharmacie du futur, c’est pas les volontaires en blouse blanche, c’est les algorithmes qui simulent des milliers de corps virtuels. Et oui, ça va faire peur aux vieux profs qui croient encore que la science se fait avec des seringues et des échantillons de sang.
Ben non, les études pharmacocinétiques c’est du bidon. Les labos trichent avec les excipients, les volontaires sont des étudiants en médecine qui ont pas d’estomac, et les normes sont fixées par des lobbyistes de Big Pharma qui veulent que les génériques restent chers.
Et puis vous avez vu la composition des génériques en Chine ? Moi j’ai comparé un générique français et un indien… c’est pas la même molécule en fait. Juste le nom.
Je suis un patient diabétique depuis 20 ans. J’ai changé trois fois de générique pour mon metformine. Aucun problème. Mais j’ai un ami qui a eu des troubles digestifs après un changement. On pense que c’est la même chose… mais le corps, il sait.
Je crois qu’on devrait avoir le droit de demander à rester sur le même générique, comme on demande un médecin ou une marque de dentifrice.
Je viens du Mali, où les génériques sont souvent la seule option. Je me suis renseigné sur ces études, et je dois dire : ça rassure. Mais je me demande… comment les pays à ressources limitées font-ils pour les tester ?
Est-ce qu’on leur impose les mêmes normes ? Ou est-ce qu’on leur dit : « Faites comme vous pouvez » ?
La bioéquivalence, c’est bien. Mais l’équité, c’est mieux.
La critique de la pharmacocinétique comme seul critère est légitime, mais elle ne doit pas servir à rejeter les génériques en bloc. Le système actuel, malgré ses limites, a sauvé des millions de vies en rendant les traitements accessibles.
La solution n’est pas de l’abandonner, mais de l’enrichir : intégrer la pharmacodynamique, les données de santé réelles, les registres de patients. La science progresse par accumulation, pas par révolution.
Je suis pharmacienne. Je vois des gens qui refusent les génériques parce qu’ils sont « moins chers ». C’est triste. Mais je comprends. On leur vend une idée : le prix = qualité.
Alors je leur dis : « Si ton générique te fait mieux marcher que ton médicament d’origine à 300€, c’est qu’il marche. »
Et je leur montre le certificat d’approbation de l’EMA. Leur regard change. C’est ça, la vraie éducation.